Version 3.70
Zika Virus Typing Tool
Tutorial
Tutorial
One or more (up to 20000 per run) Zika sequences can be pasted or uploaded in Fasta format (see Fig. 1). Sequences of all regions of the Zika genome can be used.
When pasting a single sequence the Identifier of the sequence (>Sequencename) should be included (Fasta format).
A set of example sequences is available.
The analysis takes up to a few seconds per sequence. Thus, for large batches the run can be revisited later, by typing the Job-Id at the bottom of the page. Here also runs by others can be viewed, if the Job-Id has been communicated. Runs are kept on the server for 1 week.
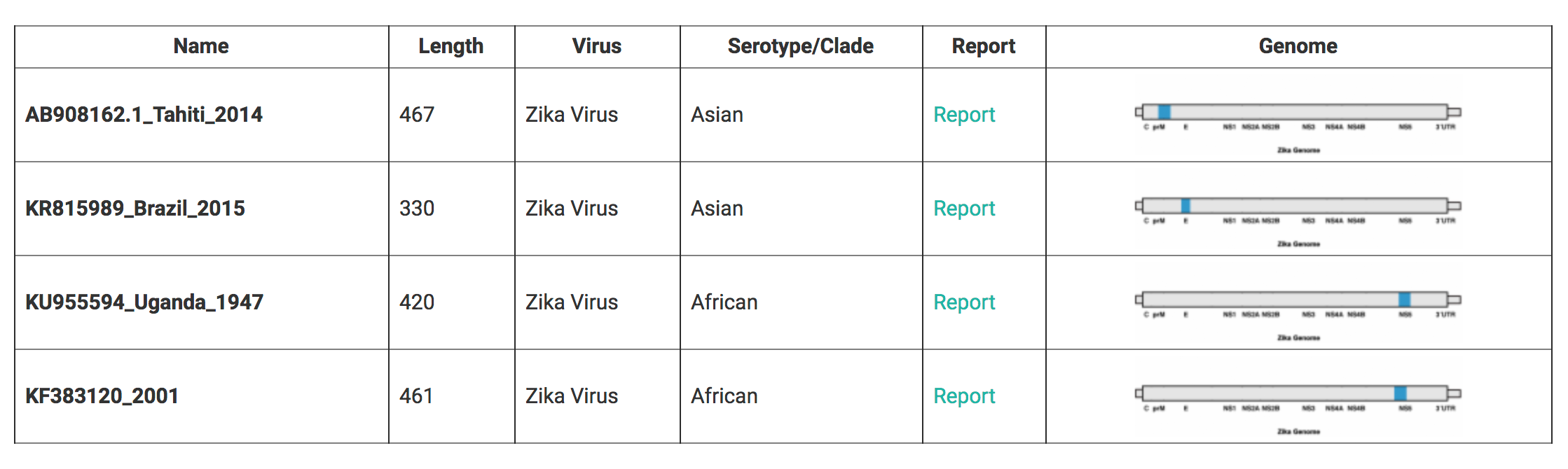
After the 'Start' button is pushed the analysis is started and after a few seconds the results overview is shown (Fig. 2). While the analysis is still in progress the following message is displayed: 'Analyzing (Showing partial results)...'.
The Job Id number, which can be used to retrieve the results later, is displayed at
the top of the page as soon as the job is started.
For each sequence the length, positioning on the Zika genome and assigned genotype
are determined.
After the complete analysis is finished the analysis results can be downloaded as
XML, CSV of Excel file.
For each sequence more details can be viewed in the individual reports by clicking
on 'Report' in the results table.
The report of the genotyping details per sequence contains a brief overview of the
assignment, followed by details of the phylogenetic analysis including a
phylogenetic tree.
The report contains information on:
- The sequence submitted (name and length)
- Blast result: the genus/species (Zika species) of the submitted sequence
- Phylo result: For Zika sequences, the genotype assignment
- The robustness of the phylogenetic assignment using bootstrap values. Only with bootstrap > 70 a genotype or variant is assigned.
- A graphical representation of the Zika genome showing the genomic region of the query sequence with the start and end positions related to the Zika reference sequence.
- A NJ tree is computed with the submitted sequence at the top
- The bootstrap tree can be viewed and downloaded in the PAUP* log file
- The alignment can be downloaded in NEXUS or FASTA format
No genotype is assigned if:
- The submitted sequence belongs to another Flavivirus genus. In this case the genus is indicated and the analysis stops.
- The submitted sequence is shorter than 200 bp. In this case only assignment of the species (Zika) is possible.
- The bootstrap value < 70. In this case an alignment and a NJ tree are produced which can be used to get an indication of the phylogeny.
